
Clinica e trattamento della distrofia muscolare di Duchenne: caso tra le malattie più frequenti delle fibre muscolari dell’età pediatrica
Nel seguente articolo si parlerà di “miopatie”: si tratta di malattie ereditarie o acquisite che colpiscono i muscoli volontari, danneggiandoli progressivamente fino a determinare un esito talvolta fatale per chi ne soffre. Esistono diverse manifestazioni cliniche di miopatie. Tra le più note vi è la distrofia muscolare di Duchenne, che sarà l’argomento centrale dell’articolo.
Cos’è la distrofia?
Nel 1954 Walton e Nattrass definirono le distrofie come “malattie primitive” (ovvero presenti alla nascita e spesso conseguenti a mutazioni genetiche) o ereditarie delle fibre muscolari. La diagnosi di distrofia è confermata da una carenza o addirittura dall’assenza della “distrofina”: si tratta di una proteina citoscheletrica presente nei muscoli striati, nei muscoli lisci e nel miocardio. Essa svolge un ruolo critico nel processo di contrazione muscolare.
I muscoli dei pazienti affetti da questo tipo di patologia vanno incontro a un progressivo depauperamento delle fibre di cui sono composti proprio a causa di un deficit della distrofina. Il tessuto muscolare è così soggetto a lacerazioni seguite dall’infiltrazione di tessuto adiposo e fibroso nella sede della lesione.
L’andamento ingravescente delle distrofie porta a una graduale perdita delle capacità motorie: i processi degenerativi non vengono sufficientemente controbilanciati dai processi riparativi e i muscoli tendono a “fibrotizzarsi”, perdendo ogni capacità di contrazione.
La distrofia muscolare di Duchenne tra le malattie pediatriche
La distrofia muscolare di Duchenne deve il suo nome a Guillame Duchenne, il neurologo francese che per primo descrisse tale patologia nel 1861. La malattia colpisce ogni anno 1 bambino su 3500 ed ha un’incidenza prettamente maschile: essendo una malattia genetica correlata al cromosoma X le femmine possono essere portatrici sane, ma non manifestano sintomi.
La diagnosi è clinica, ma può essere confermata da test di laboratorio come gli esami ematochimici (dosaggio dell’enzima CK) e l’elettromiografia, la quale prevede l’inserimento di un elettrodo ad ago nel muscolo per misurarne l’attività elettrica.
L’ultima parola spetta alla biopsia muscolare: si tratta di un esame mininvasivo che consente di verificare l’assenza della distrofina e la presenza di infiltrazioni di tessuto connettivo nel muscolo , confermando in questo modo la diagnosi.
È anche possibile eseguire uno screening pre-natale dopo la decima settimana di vita intrauterina attraverso un esame di genetica molecolare.
La clinica della distrofia tra le malattie dei bambini
La distrofia muscolare di Duchenne è considerata una malattia pediatrica: viene infatti diagnosticata intorno al terzo-quarto anno di vita.
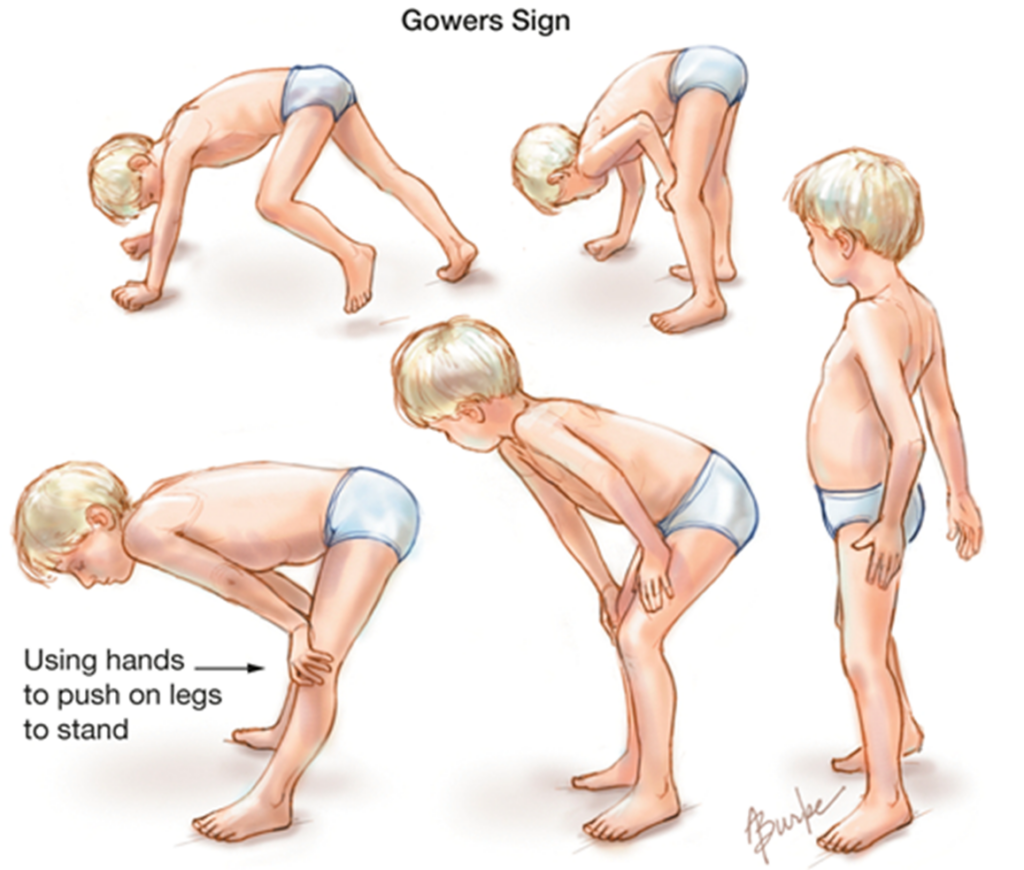
Il sintomo iniziale con cui la patologia si manifesta è il “Segno di Gowers”: si tratta di un movimento caratteristico di quei bambini che faticano ad assumere la posizione eretta, per cui necessitano di aiutarsi appoggiandosi a supporti circostanti o facendo leva con le mani sulle proprie gambe per alzarsi in piedi. Talvolta si tende a sottostimare il fenomeno definendo il bambino semplicemente come “pigro”.
Un altro sintomo che desta sospetti è il ritardo nell’inizio della deambulazione. La camminata del bambino appare inoltre goffa ed incerta, talvolta eseguita in punta di piedi e caratterizzata dalla difficoltà ad assumere un’andatura veloce o a fare le scale.
Vi è poi un ulteriore segno che, se presente in concomitanza a quelli appena descritti, rappresenta un vero e proprio campanello d’allarme: si tratta della pseudo-ipertrofia dei polpacci causata dalla sostituzione del tessuto muscolare con tessuto fibrotico.
Qualora si assistesse alla compresenza di questi fattori, potrebbe essere il caso di rivolgersi a un neurologo per giungere il più precocemente possibile a una diagnosi sicura.
Decorso della malattia
La distrofia muscolare di Duchenne si caratterizza per una progressione lenta e ingravescente che solitamente comporta la perdita di autonomia motoria tra i 7 e i 12 anni di età.
Il primo segno evidente, come già evidenziato, è la deambulazione difficoltosa caratterizzata da frequenti cadute. Verso i 5-6 anni cominciano a diventare più evidenti i deficit di forza a livello degli arti superiori e degli arti inferiori, associati allo sviluppo di retrazioni muscolo-tendinee e scoliosi. Solitamente il paziente perde la capacità di camminare entro i 12 anni.
Una complicazione a cui quasi tutti i pazienti vanno incontro è lo sviluppo di gravi problematiche cardiache e respiratorie: un paziente su cinque muore a causa di gravi cardiomiopatie secondarie alla distrofia.
L’aspettativa di vita, in ogni caso, non supera la terza o la quarta decade di età.
Trattamento e cura
Al giorno d’oggi non esiste un trattamento risolutivo per le miopatie degenerative ad esordio precoce; tuttavia, la ricerca sta facendo passi da gigante.
La presa in carico del paziente da parte di un équipe multidisciplinare di esperti ha permesso un notevole miglioramento delle aspettative di sopravvivenza dei pazienti affetti.
L’adeguato trattamento farmacologico associato a fisioterapia, chirurgia ortopedica, monitoraggio cardiologico e assistenza respiratoria si è già dimostrato efficace nel ridurre i sintomi e migliorare l’aspettativa e la qualità di vita.
Il trattamento fisioterapico rivolto alla gestione delle retrazioni muscolo tendinee e al controllo del dolore dei vari distretti articolari può essere utile per prevenire l’ipotrofia e migliorare la forza muscolare. Il fisioterapista svolge anche un ruolo importante nell’educazione all’utilizzo di ausili e ortesi utili per contenere le deformità muscoloscheletriche causate dal peggioramento della malattia e che consentono di recuperare una certa autonomia nelle attività della vita quotidiana.
Non ultimo per importanza, Il supporto psicologico ed emotivo fornito da personale esperto e qualificato può essere determinante nella convivenza con la malattia, tanto per il paziente quanto per i famgiliari su cui grava un grande carico assistenziale.
Marco Manzoni per Questione Civile
Sitografia
Birnkrant, D. J., Bushby, K., Bann, C. M., Alman, B. A., Apkon, S. D., Blackwell, A., Case, L. E., Cripe, L., Hadjiyannakis, S., Olson, A. K., Sheehan, D. W., Bolen, J., Weber, D. R., & Ward, L. M. (2018). Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. The Lancet. Neurology, 17(4), 347–361. https://doi.org/10.1016/S1474-4422(18)30025-5
Duan, D., Goemans, N., Takeda, S., Mercuri, E., & Aartsma-Rus, A. (2021). Duchenne muscular dystrophy. Nature Reviews. Disease Primers, 7(1). https://doi.org/10.1038/S41572-021-00248-3
Sun, C., Shen, L., Zhang, Z., & Xie, X. (2020). Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes, 11(8), 1–25. https://doi.org/10.3390/GENES11080837
